
3-A.実用的な量子分子動力学プログラムの開発
DCFTBMD
分子動力学(MD)シミュレーションは、複雑系が示す現象や機能を、時々刻々と変化する原子の運動としてとらえる手法である。そのため、物理・化学・生命科学・材料科学・工学など様々の分野で用いられている。MDシミュレーションは原子に働く力に基づいて運動方程式を数値的に解く方法であるが、通常、経験的な分子力場が用いられる。一方、第一原理計算や量子化学計算などと組み合わせてon-the-flyに運動方程式を解く方法も行われるようになってきた。前者を古典MD(CMD)法、後者を量子MD(QMD)法と区別する場合もある。CMD法は、経験的な分子力場では化学結合の生成・開裂を表現することが困難なので、CMD法では化学反応を取り扱うことが難しい。一方、QMD法は、任意の原子配置に対する電子状態を求めるので、(計算精度の差こそあるが)化学反応を取り扱うことができる。しかしながら、QMD法はCMD法に比べて、第一原理計算や量子化学計算がボトルネックとなり、取り扱える系の大きさ、すなわち、原子数が制限される。実際、CMD法では数万原子系・数ナノ秒のシミュレーションが研究室のワークステーションを用いても可能であるが、QMD法ではスーパーコンピュータを用いても数百原子系・数ピコ秒のシミュレーションが限界であった。このCMD法とQMD法の大きなギャップを埋めるべく、中井研究室では分割統治型密度汎関数強束縛(DC-DFTB)法を用いたMDシミュレーション手法、すなわち、DC-DFTB-MD法を開発してきた。さらに、超並列環境に適したプログラムDCDFTBMDを一から開発してきた。このプログラムを用いて、数多くの応用研究を実施してきた(図3-A-1)。これらの応用研究を通して、DCDFTBMDプログラムが世界的にも類を見ない性能を有することを確証できたので、2018年11月より公開を開始した。DCDFTBMDプログラムのユーザー登録は専用ページから行うことができる。DCDFTBMDプログラムの機能については、英語解説記事(J. Comput. Chem., 40, 1538 (2019))および日本語解説記事(J. Comput. Chem. Jpn., 17, A21 (2018))を参考にしていただきたい。DCDFTBMDプログラムの機能および性能は現在も改良中である。最近では、スーパーコンピュータ「富岳」を用いて、1点計算であれば1億原子系も取り扱えるようになった。また、後述する励起状態ダイナミクスは、QMD法の特徴を最大限に生かせる注目すべき展開である。
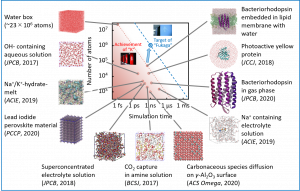
図3-A-1
ここでは、DCDFTBMDプログラムを用いた応用研究をいくつか紹介する。最初に行った研究は、水中におけるプロトン拡散である。通常、水中のプロトンはヒドロニウム(H3O+)イオンとして存在し、H3O+イオンのまま拡散するビークル(vehicule)機構と水素結合ネットワークを通してプロトンが移動するグロータス(Grotthuss)機構が知られている。Grotthuss拡散はまさに結合の開裂と生成を繰り返して起こる現象であり、通常のCMDシミュレーションでは取り扱いが困難であり、特別な分子力場が必要となる。また、水中のでGrotthuss拡散はが起こる場所の限定が困難であるため、量子力学的に取り扱う領域を限定するQM-MM/MDシミュレーションも困難となる。我々は全系を量子力学的に取り扱うDC-DFTB-MD法により、vehicle機構およびGrotthuss機構の拡散係数を見積り、実験値とかなり良い一致をすることを確認した。さらに、拡散係数の温度依存性からアーレニウスプロットにより拡散障壁も見積り、これまた実験値を再現することを確認した。この成功を受けて、氷中のプロトン拡散についてもDC-DFTB-MDシミュレーションを行った(図3-A-2)。そして、氷中ではvehicle拡散はほとんど起こらないが、構造化された相ほどGrotthuss拡散が起こりやすいことがわかった。
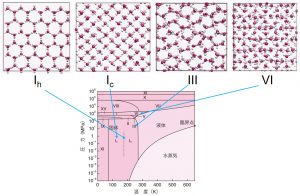
図3-A-2
このシミュレーション結果を北川宏教授(京都大学)にお見せしたところ、北川グループで合成したナノチューブ中の水クラスターが高いプロトン伝導性を示すことをご教授いただいた。そこで、この系におけるプロトン拡散についてもDC-DFTB-MDシミュレーションを行い、常温でも構造化された水クラスターによりGrotthuss拡散を容易にしていることを明らかにした(図3-A-3)。
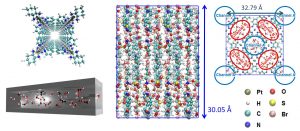
図3-A-3
水中の水酸化物イオン(OH–)の拡散についても、DC-DFTB-MDシミュレーションを行った。そして、vehicle拡散に加えて、水素結合ネットワークを介したプロトン移動によるGrotthuss拡散が起こることもわかった。この結果は、CO2化学吸収法の研究でも重要な知見となった。中井研究室では、企業・実験研究者からなるチームに加わり、2009年より約10年間、CO2分離回収技術の一つであるCO2化学吸収法の研究を行ってきた(図3-A-4)。そして、DC-DFTB-MD法が開発できるとすぐにこの系に応用した。そして、巨視的には化学反応の正反応と逆反応であるが、微視的には異なる機構で反応が進行していることを明らかにした(図3-A-5)。低温で起こる正反応では、水酸化物イオンがGrotthuss拡散により、水溶液中のCO2分子あるいは双性イオンにアタックし、重炭酸イオン(HCO3–)またはカルバメートを生成する。一方、高温で起こる逆反応では、負電荷を帯びた重炭酸イオンまたはカルバメートと正電荷を帯びたプロトン化アミンが直接相互作用したイオン対を形成して、イオン対間のプロトン移動によりCO2分子が放出される。
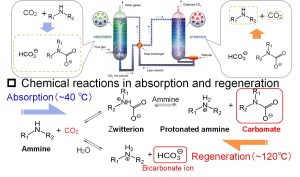
図3-A-4
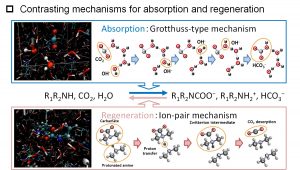
図3-A-5
中井研究室では、生命科学分野にもDC-DFTB-MDシミュレーションを応用している。その対象の一つがバクテリオロドプシン(BR)である。BRは、光駆動プロトンポンプとしてエネルギー変換を行う膜タンパク質であり、光エネルギーによって細胞内のプロトンを細胞外に能動輸送する。その後、濃度勾配をプロトン駆動力として利用することで、FoF1-ATP合成酵素はADPとリン酸からATPを合成する。BRによるプロトンポンプには、少なくとも5段階のプロトン移動が起こっていることが実験的に示唆されている。2016年には、X線自由電子レーザー(XFEL)により、光サイクルにおける13個構造のスナップショットが報告された(図3-A-6)。
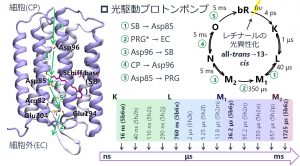
図3-A-6
我々はまず、第1段階目のプロトン移動に関する研究に着手し、BR全系および内部水分子を含めた~3,800原子系を用いたDC-DFTB-メタダイナミクス(MTD)シミュレーションを行った。シッフ塩基(SB)上のプロトンが水分子を介してアスパラギン酸(Asp85)に移動する機構(図3-A-7(左))と、Asp85が水分子からプロトンを引き抜き、OH–を生成した後、SBからOH–にプロトン移動する機構(図3-A-7(右))の両方について自由エネルギー面を求めた。その結果、後者のOH–を介してプロトン移動する自由エネルギー障壁の方が低いことが明らかとなった。また、この第1段階のプロトン移動はL型中間体のうち、760 ns(5b6x)または2 μs(5h2k)で起こることも明らかにした。
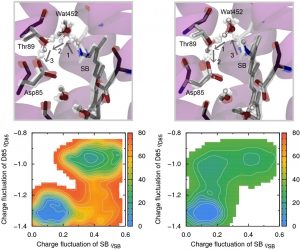
図3-A-7
第2,3段階目のプロトン移動に関する研究では、BR全系、内部水分子、脂質二重膜、そして、水溶媒を含めた~50,000原子系のDC-DFTB-MD/MTDシミュレーションを行った(図3-A-8)。
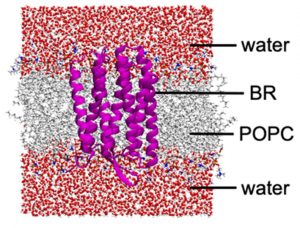
図3-A-8
中井研究室では、リチウムイオン二次電池(LIB)、そしてポストLIBとしてナトリウムイオン二次電池(SIB)、カリウムイオン二次電池(PIB)に関する研究を、山田淳夫教授(東京大学)との共同研究として進めている。超濃厚電解液は、高酸化還元耐性、化学的・熱的安定性、低揮発性という特長をもつことが実験的に報告された。また、電解液の粘性が上がるにもかかわらず、キャリア移動度があまり低下しないことも報告された。DC-DFTB-MDシミュレーションの結果、濃厚化に伴いキャリアイオンがアニオンを介してつながった凝集状態となり、拡散能は低下するが、配位子交換によりキャリアイオンが輸送されることが示された。高電位水系SIB/PIBでは、濃厚化によりすべての水分子がカチオンに溶媒和したハイドレートメルト状態が形成され、その結果、HOMO準位の低下がもたらされ、結果として広い電位窓が導かれることが示された(図3-A-9)。

図3-A-9
DFTBはDFTに強束縛近似を用いて効率化を図った半経験法である。最近、励起状態を計算する手法として、TDDFTを近似したTDDFTBが提案された。中井研究室では、励起状態ダイナミクスシミュレーションを実行するために、DCDFTBMDプログラムにTDDFTBに基づくエネルギーおよびエネルギー勾配の計算コードを実装した。さらに、長距離補正(LC-TDDFTB)の導入やスピン反転(SF-TDDFTB)への拡張も行った。DC法(DC-TDDFTB)による効率化も実装した。励起状態は一般に多くの状態が密接に存在するため、励起状態ダイナミクスには状態間遷移を考慮する必要がある。そこで、Tully博士によって提案された最小交換サーフィスホッピング(FSSH)を導入した。このTDDFTB-FSSHにより、ペロブスカイト太陽電池(PSC)における光励起に伴う電荷分離過程のシミュレーションに成功した(図3-A-10)。現実に近いモデルを用いた励起状態ダイナミクスシミュレーションを実行できるのは、DCDFTBMDプログラムのセールスポイントと考える。今後、様々な系に応用していく予定である。
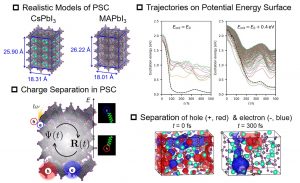
図3-A-10
重要文献
<DC-DFTB-MD>
- H. Nishizawa, Y. Nishimura, M. Kobayashi, S. Irle, H. Nakai, “Three pillars for achieving quantum mechanical molecular dynamics simulations of huge systems: Divide-and-conquer, density functional tight-binding, and massively parallel computation”, J. Comput. Chem., 37, 1983 (2016).
- Y. Nishimura, H. Nakai, “Hierarchical parallelization of divide-and-conquer density functional tight-binding molecular dynamics and metadynamics simulations”, J. Comput. Chem., 41, 17592 (2020).
- Y. Nishimura, H. Nakai, “Quantum chemical calculations for up to one hundred million atoms using DCDFTBMD code on supercomputer Fugaku”, Chem. Lett., in press (2021).
<DCDFTBMD program>
- Y. Nishimura, H. Nakai, “DCDFTBMD: Divide-and-conquer density functional tight-binding program for huge-system quantum mechanical molecular dynamics simulations”, J. Comput. Chem., 40, 1538 (2019).
<Proton Diffusion>
- H. Nakai, A. W. Sakti, Y. Nishimura, “Divide-and-conquer-type density-functional tight-binding simulations of proton diffusion in a bulk water system”, J. Phys. Chem. B, 120, 217 (2016).
- A. Sakti, Y. Nishimura, H. Nakai, “Divide-and-conquer-type density-functional tight-binding simulations of hydroxide ion diffusion in bulk water”, J. Phys. Chem. B, 121, 1362 (2017).
- A. Sakti, Y. Nishimura, C. Chou, H. Nakai, “Density-functional tight-binding molecular dynamics simulations of excess proton diffusion in ice Ih, Ice Ic, Ice III, and melted ice VI phases”, J. Phys. Chem. A, 122, 33 (2018).
- K. Otake, K. Otsubo, T. Komatsu, S. Dekura, J. M. Taylor, R. Ikeda, K. Sugimoto, A. Fujiwara, Y. Nanba, T. Ishimoto, M. Koyama, C. Chou, A. Sakti, Y. Nishimura, H. Nakai, H. Kitagawa, “Confined water-mediated high proton conduction in hydrophobic channel of a synthetic nanotube”, Nature Commun., 11, 843 (2020).
<CO2 Chemical Absorption>
- H. Nakai, Y. Nishimura, T. Kaiho, T. Kubota, H. Sato, “Contrasting mechanisms for CO2 absorption and regeneration processes in aqueous amine solutions: Insights from density-functional tight-binding molecular dynamics simulations”, Chem. Phys. Lett., 647, 127 (2016).
- A. Sakti, Y. Nishimura, H. Sato, H. Nakai, “Divide-and-conquer density-functional tight-binding molecular dynamics study on the formation of carbamate ions during CO2 chemical absorption in amine solutions”, Bull. Chem. Soc. Jpn., 90, 1230 (2017).
- A. Sakti, Y. Nishimura, H. Nakai, “Rigorous pKa estimation of amine species using density-functional tight-binding-based metadynamics simulations”, J. Chem. Theory Comput., 14, 351 (2018).
<BR>
- J. Ono, M. Imai, Y. Nishimura, H. Nakai, “Hydroxide ion carrier of proton pumps in bacteriorhodopsin: Primary proton transfer”, J. Phys. Chem. B, 124, 8524 (2020).
<LIB/SIB/PIB>
- M. Okoshi, C.-P. Chou, H. Nakai, “Theoretical analysis of carrier ion diffusion in superconcentrated electrolyte solutions for sodium-ion batteries”, J. Phys. Chem. B, 122, 2600 (2018).
- K. Doi, Y. Yamada, M. Okoshi, J. Ono, C-P. Chou, H. Nakai, A. Yamada, “Reversible sodium metal electrodes: Is fluorine an essential interphasial component?”, Angew. Chem. Int. Ed., 58, 8024 (2019).
- Q. Zheng, S. Miura, S. Ko, K. Miyazaki, E. Watanabe, M. Okoshi, C.-P. Chou, Y. Nishimura, H. Nakai, T. Kamiya, T. Honda, J. Akikusa, Y. Yamada, A. Yamada, “Sodium- and potassium-hydrate melts containing asymmetric imide anions for high-voltage aqueous batteries”, Angew. Chem. Int. Ed., 58, 14202 (2019).
<TDDFTB-MD>
- N. Komoto, T. Yoshikawa, J. Ono, Y. Nishimura, H. Nakai, “Development of large-scale excited-state calculations based on the divide-and-conquer time-dependent density functional tight-binding method”, J. Chem. Theory Comput., 15,1719 (2019).
- N. Komoto, T. Yoshikawa, Y. Nishimura, H. Nakai, “Large-scale molecular dynamics simulation for ground and excited states based on divide-and-conquer long-range corrected density functional tight-binding method”, J. Chem. Theory Comput., 16, 2369 (2020).
<SF-TDDFTB>
- M. Inamori, T. Yoshikawa, Y. Ikabata, Y. Nishimura, H. Nakai, “Spin-flip time-dependent density-functional tight-binding method for application to conical intersection”, J. Comput. Chem., 41, 1538 (2020).
<TDDFTB-FSSH>
- H. Uratani, H. Nakai, “Non-adiabatic molecular dynamics with divide-and-conquer type large-scale excited state calculations”, J. Chem. Phys., 152, 224109 (2020).
- H. Uratani, T. Morioka, T. Yoshikawa, H. Nakai, “Fast nonadiabatic molecular dynamics via spin-flip time-dependent density-functional tight-binding approach: Application to nonradiative relaxation of tetraphenylethylene with locked rings”, J. Chem. Theory Comput., 16, 7299 (2020).
- H. Uratani, T. Yoshikawa, H. Nakai, “Trajectory surface hopping approach to condensed-phase nonradiative relaxation dynamics using divide-and-conquer type spin-flip time-dependent density-functional tight-binding”, J. Chem. Theory Comput., 17, 1290 (2021).
<Perovskite Solar Cell>
- H. Uratani, C-P. Chou, H. Nakai, “Quantum mechanical molecular dynamics simulations of polaron formation in methylammonium lead iodide perovskite”, Phys. Chem. Chem. Phys., 22, 97 (2020).
- H. Uratani, H. Nakai, “Simulating the coupled structural–electronic dynamics of photo-excited lead iodide perovskites”, J. Phys. Chem. Lett., 11, 4448 (2020).
<Review>
- A. Sakti, Y. Nishimura, H. Nakai, “Recent Advances in Quantum-Mechanical Molecular Dynamics Simulations of Proton Transfer Mechanism in Various Water-Based Environments”, WIREs Comput. Mol. Sci., 10, e1419 (2019).
- C. Chou, A. Sakti, Y. Nishimura, H. Nakai, “Development of divide-and-conquer density-functional tight-binding method for theoretical research on Li-ion battery”, Chem. Rec., 18, 746 (2019).
<日本語解説>
- 西村 好史, 吉川 武司, 中井 浩巳, “DCDFTBMDプログラムの公開”, J. Comput. Chem. Jpn., 17, A21 (2018).