
3-C.Implementation to de facto standard quantum chemistry program
GAMESS
The GAMESS program is one of the most widely used quantum chemistry programs developed and managed by Prof. Mark Gordon (Iowa State University). Since the source code of the GAMESS program is also provided, Nakai’s Group has used it as a platform for theoretical development. All the methods introduced in “1. Development of a practical theoretical calculation method that can correctly reproduce a real system” have benefited from the GAMESS program in some way. From the viewpoint of “Social Implementation of Computational Chemistry”, we have implemented some of our theoretical methods in the public version of the GAMESS program.
The first published method is the divide-and-conquer (DC) method. In November 2008, with the cooperation of Dr. Mike Schmidt, the implementation work was carried out at Iowa State University by Dr. Masato Kobayashi (currently Hokkaido University). The DC-HF/DFT method and various DC-correlation methods were implemented, supporting not only energy calculation but also structure optimization calculation. The users need to flag (DCFLG = .TRUE., DODCCR = .TRUE.) in the corresponding namelist ($DANDC, $DCCORR). For the other parameters, default values are available (Fig. 3-C-1). In addition, since the DC method can break any chemical bonds, the users only need to specify the automatic partitioning option (SUBTYPE = AUTO) to determine subsystems. Unlike other fragmentation-type linear-scaling methods, the DC method can treat fractional numbers of electrons and spins of the subsystems, which the users consequently need not to specify.
The DC-HF/DFT calculations are faster than the conventional HF/DFT ones only for hundreds of atomic systems or more. On the other hand, DC-correlation calculations are sufficiently effective in dozens of atomic systems. Although it is not well known, the DC-correlation calculations do not necessarily require the DC-HF calculations. Namely only the correlation calculations using the DC method can be performed after performing the standard HF calculations.
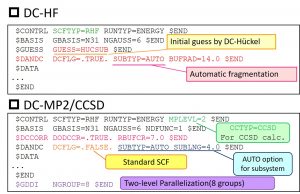
Fig. 3-C-1
The next implemented method is the local response dispersion (LRD) method. In March 2011, with the cooperation of Dr. Mike Schmidt, the implementation work was carried out at Iowa State University by Dr. Yasuhiro Ikabata (currently Toyohashi University of Technology). To use LRD for DFT dispersion correction, the users flag (LRDFLG = .TRUE.) in the corresponding name list ($DFT). When the LC-BOP-LRD functional is selected (DFTTYP = LCBOPLRD), the flag is set automatically.
The third implemented method is the local unitary transformation (LUT) method. In June 2015, with the cooperation of Dr. Mike Schmidt, the implementation work was carried out at Waseda University by Dr. Yuya Nakajima. Since the implementation was limited to the range using real-type variables used in GAMESS, only the spin-free one-component relativistic method was introduced. Also, only the LUT method for one electron term was implemented. Nevertheless, its performance is reasonably high. The official manual highly evaluated our implementation (Fig. 3-C-2). Currently, we are also implementing the LUT method for two-electron terms.

Fig. 3-C-2
Key Literature
<DC-in-GAMESS(in Japanese)>
- M. Kobayashi, T. Akama, H. Nakai, “分割統治(DC)電子状態計算プログラムのGAMESSへの実装”, J. Comput. Chem. Jpn., 8, 1 (2009).
<LUT-in-GAMESS>
- Y. Nakajima, J. Seino, M. W. Schmidt, H. Nakai, “Implementation of efficient two-component relativistic method using local unitary transformation to GAMESS program”, J. Comput. Chem. Jpn., 15, 68 (2016).
- C. Takashima, J. Seino, H. Nakai, “Implementation of picture change corrected density functional theory based on infinite-order two-component method to GAMESS program”, J. Comput. Chem. Jpn., 19, 128 (2020).