
3-A.Development of practical quantum molecular dynamics program
DCFTBMD
Molecular dynamics (MD) simulation is a method of describing the phenomena and functions of complex systems as the time-course change of atomic motions, and therefore, it is used in various fields such as physics, chemistry, biology, material science, and engineering. Classical MD (CMD) simulations use an empirical molecular force field to numerically solve the equation of motion based on the force acting on an atom. On the other hand, quantum MD (QMD) simulations solve the equation of motion on-the-fly in combination with quantum-chemistry or first-principles calculations. Since the CMD method is difficult to treat chemical reactions because of less description of the formation and cleavage of chemical bonds by using the empirical molecular force field. On the other hand, the QMD method can demonstrate chemical reactions using the atomic forces based on the electronic state for an arbitrary atomic configuration. However, compared to the CMD method, the QMD method has a bottleneck in quantum-chemistry/first-principles calculations, of which the computational time increases rapidly as a larger system size. In fact, the CMD simulations can be performed for tens of thousands of atomic systems and several nanoseconds using a workstation in the laboratory, but the QMD simulations are limited to hundreds of atomic systems and several picoseconds even by using a supercomputer.
In order to fill the large gap between the CMD and QMD methods, Nakai’s Group has developed an MD simulation technique using the divide-and-conquer density-functional tight-binding (DC-DFTB) method, that is, DC-DFTB-MD. Furthermore, we have developed a program DCDFTBMD suitable for massively parallel environments from scratch. Various application studies have been carried out using this program (Fig. 3-A-1). Through such applications, we were able to confirm that the DCDFTBMD program has unparalleled performance in the world, so we started releasing the program in November 2018. User registration for the DCDFTBMD program can be done from our web site. For the functions of the DCDFTBMD program, see the English commentary article (J. Comput. Chem., 40, 1538 (2019)) and the Review (in Japanese) article (J. Comput. Chem. Jpn., 17, A21 (2018)). The functionality and performance of the DCDFTBMD program is still being improved. Recently, using the supercomputer “FUGAKU”, we could perform the electronic structure calculation for a 100 million atomic systems with the DCDFTBMD program. Excited-state dynamics, which will be described later, is a remarkable development that makes the best use of the features of the QMD method.
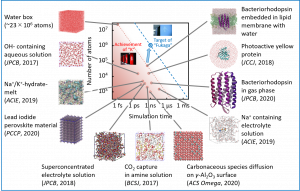
Fig. 3-A-1
Let us explain here some application studies using the DCDFTBMD program. The first study was related with proton diffusion in water. While protons in water normally exist as hydronium (H3O+) ions, two proton diffusion mechanisms are reported: namely, the vehicle mechanism that diffuses H3O+ ions as they are and the Grotthuss mechanism that protons move through a hydrogen bond network. Because Grotthuss diffusion is a phenomenon that occurs by repeating the cleavage and formation of chemical bonds, the CMD method is difficult to treat it and requires a special force field. In addition, since Grotthuss diffusion occurs ubiquitously in water, QM-MM/MD simulations that limit the region handled quantum mechanically are difficult to be applied. We could evaluated the diffusion constants of the vehicle and Grotthuss mechanisms with a reasonable accuracy by the DC-DFTB-MD method, which treats the entire system quantum mechanically. Furthermore, the diffusion barrier, which was estimated by the Arrhenius plot from the temperature dependence of the diffusion coefficient, could reproduce the experimental value well. Following this success, DC-DFTB-MD simulations were also performed for proton diffusion in ice (Fig. 3-A-2). The simulations clarified that Grotthuss diffusion is more likely to occur in the structured ice phase, although vehicle diffusion rarely occurs.
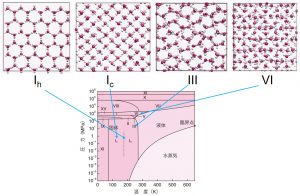
Fig. 3-A-2
Prof. Hiroshi Kitagawa (Kyoto University) informed us that the water clusters in the nanotubes synthesized by his group exhibit high proton conductivity. We performed DC-DFTB-MD simulations of proton diffusion in this system and found that structured water clusters facilitate Grotthuss diffusion even at room temperature (Fig. 3-A-3).
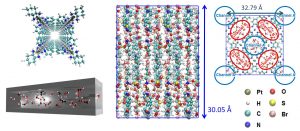
Fig. 3-A-3
DC-DFTB-MD simulations were also performed for the diffusion of hydroxide ions (OH–) in water. In addition to vehicle diffusion, Grotthuss diffusion was found to occur due to proton transfer via a hydrogen bond network. The result was an important finding in the study of CO2 chemical absorption method, which is one of the CO2 capture and separation technologies (Fig. 3-A-4). Nakai’s Group have been conducting the research on the CO2 chemical absorption method for about 10 years since 2009, as a collaborative study with company and experimental researchers. The DC-DFTB-MD simulations clarified that the reactions in the absorption and regeneration towers are proceeding by two different mechanisms from the microscopic point of view, although they correspond to forward and backward reactions macroscopically (Fig. 3-A-5). In the forward reaction that occurs at low temperatures, hydroxide ions attack CO2 molecules and zwitterions in aqueous solution by Grotthuss diffusion to produce bicarbonate (HCO3–) and carbamate ions, respectively. On the other hand, in the backward reaction that occurs at high temperature, negatively charged bicarbonate or carbamate ion and positively charged protonated amine form an ion pair that directly interacts, and releases CO2 molecule by the proton transfer between the ion pairs.
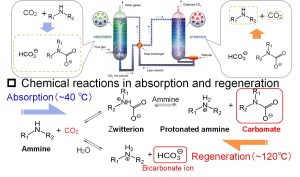
Fig. 3-A-4
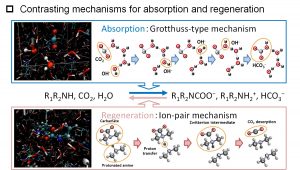
Fig. 3-A-5
Nakai’s Group is also applying DC-DFTB-MD simulations to the field of biology. One of the targets is bacteriorhodopsin (BR), which is a membrane protein that performs energy conversion as a light-driven proton pump, and actively transports intracellular protons to the outside of the cell by light energy. The FoF1-ATP synthase then synthesizes ATP from ADP and phosphate by using the concentration gradient as a proton driving force. Experimental studies reported that the proton pump by BR has at least 5 stages of proton transfer. In 2016, an X-ray free-electron laser (XFEL) study reported snapshots of 13 structures in the optical cycle (Fig. 3-A-6).
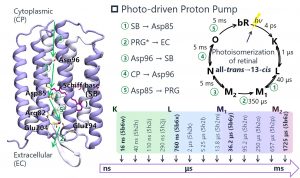
Fig. 3-A-6
We first embarked on a study on the primary proton transfer and performed DC-DFTB-metadynamics (MTD) simulations using the entire BR system including internal water molecules (~3,800 atoms). Free energy surfaces for two different mechanisms: namely, one is that a proton on the Schiff base (SB) moves to aspartic acid (Asp85) via a water molecule (Fig. 3-A-7 (left)), and the other is that Asp85 abstracts a proton from a water molecule to generate OH– and another proton of SB transfers to OH– (Fig. 3-A-7 (right)). The DC-DFTB-MTD simulations clarified that the free energy barrier via OH– is lower and that the primary proton transfer occurs in the time range of 760 ns (5b6x) or 2 μs (5h2k) of L-type intermediates.
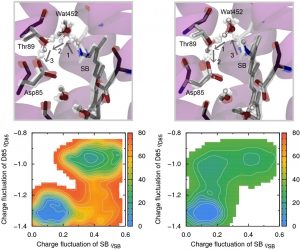
Fig. 3-A-7
In the studies on the secondary and tertiary proton transfers, DC-DFTB-MD/MTD simulations with ~50,000 atomic systems including the entire BR system, internal water molecules, lipid bilayer, and water solvent were being performed (Fig. 3-A-8).
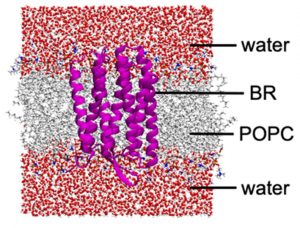
Fig. 3-A-8
Nakai’s Group are promoting collaborative studies with Prof. Atsuo Yamada (Univ. of Tokyo) on lithium-ion secondary batteries (LIB), sodium-ion secondary batteries (SIB), and potassium-ion secondary batteries (PIB).
Experimental studies reported that the highly concentrated electrolyte has the features of high redox resistance, chemical/thermal stability, and low volatility. Furthermore, the carrier mobility did not decrease so much even though the viscosity of the electrolytic solution increased. DC-DFTB-MD simulations clarified that carrier ions are transported by ligand exchange, although the diffusivity is reduced due to the aggregated state, in which carrier ions are connected via anions as the concentration increases.
Experimental and theoretical collaborations on high-voltage aqueous SIB/PIB clarified that highly concentrated Na or K aqueous electrolyte forms a hydrate melt state, in which all water molecules are solvated with cations, leading a lower HOMO level, which results in a wider electrochemical window (Fig. 3-A-9).

Fig. 3-A-9
DFTB is a semi-empirical method that uses tight binding approximation for DFT to improve efficiency. Recently, TDDFT B, which approximates TDDFT, has been proposed as a method for calculating the excited state. Nakai’s Group implemented the TDDFTB-based energy and energy gradient calculation code in the DCDFTBMD program to perform excited-state dynamics simulations. In addition, we improved the TDDFTB method by introducing long-range correction (LC-TDDFTB) and extending to spin-flip formula (SF-TDDFTB). Efficiency improvement by combining with the DC method (DC-TDDFTB) was also carried out. Since many states are generally closely present in the excited state, the transitions between states should be taken into account in the excited-state dynamics. Therefore, we introduced the fewest switched surface hopping (FSSH) proposed by Dr. Tully. TDDFTB-FSSH simulations succeeded in demonstrating the charge separation process associated with photoexcitation in a perovskite solar cell (PSC) (Fig. 3-A-10). The capability of DCDFTBMD program to perform excited-state dynamics simulations using more realistic models is considered to be the selling point.
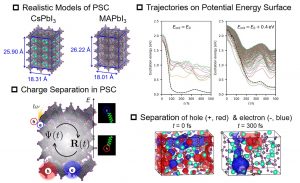
Fig. 3-A-10
Key Literature
<DC-DFTB-MD>
- H. Nishizawa, Y. Nishimura, M. Kobayashi, S. Irle, H. Nakai, “Three pillars for achieving quantum mechanical molecular dynamics simulations of huge systems: Divide-and-conquer, density functional tight-binding, and massively parallel computation”, J. Comput. Chem., 37, 1983 (2016).
- Y. Nishimura, H. Nakai, “Hierarchical parallelization of divide-and-conquer density functional tight-binding molecular dynamics and metadynamics simulations”, J. Comput. Chem., 41, 17592 (2020).
- Y. Nishimura, H. Nakai, “Quantum chemical calculations for up to one hundred million atoms using DCDFTBMD code on supercomputer Fugaku”, Chem. Lett., in press (2021).
<DCDFTBMD program>
- Y. Nishimura, H. Nakai, “DCDFTBMD: Divide-and-conquer density functional tight-binding program for huge-system quantum mechanical molecular dynamics simulations”, J. Comput. Chem., 40, 1538 (2019).
<Proton Diffusion>
- H. Nakai, A. W. Sakti, Y. Nishimura, “Divide-and-conquer-type density-functional tight-binding simulations of proton diffusion in a bulk water system”, J. Phys. Chem. B, 120, 217 (2016).
- A. Sakti, Y. Nishimura, H. Nakai, “Divide-and-conquer-type density-functional tight-binding simulations of hydroxide ion diffusion in bulk water”, J. Phys. Chem. B, 121, 1362 (2017).
- A. Sakti, Y. Nishimura, C. Chou, H. Nakai, “Density-functional tight-binding molecular dynamics simulations of excess proton diffusion in ice Ih, Ice Ic, Ice III, and melted ice VI phases”, J. Phys. Chem. A, 122, 33 (2018).
- K. Otake, K. Otsubo, T. Komatsu, S. Dekura, J. M. Taylor, R. Ikeda, K. Sugimoto, A. Fujiwara, Y. Nanba, T. Ishimoto, M. Koyama, C. Chou, A. Sakti, Y. Nishimura, H. Nakai, H. Kitagawa, “Confined water-mediated high proton conduction in hydrophobic channel of a synthetic nanotube”, Nature Commun., 11, 843 (2020).
<CO2 Chemical Absorption>
- H. Nakai, Y. Nishimura, T. Kaiho, T. Kubota, H. Sato, “Contrasting mechanisms for CO2 absorption and regeneration processes in aqueous amine solutions: Insights from density-functional tight-binding molecular dynamics simulations”, Chem. Phys. Lett., 647, 127 (2016).
- A. Sakti, Y. Nishimura, H. Sato, H. Nakai, “Divide-and-conquer density-functional tight-binding molecular dynamics study on the formation of carbamate ions during CO2 chemical absorption in amine solutions”, Bull. Chem. Soc. Jpn., 90, 1230 (2017).
- A. Sakti, Y. Nishimura, H. Nakai, “Rigorous pKa estimation of amine species using density-functional tight-binding-based metadynamics simulations”, J. Chem. Theory Comput., 14, 351 (2018).
<BR>
- J. Ono, M. Imai, Y. Nishimura, H. Nakai, “Hydroxide ion carrier of proton pumps in bacteriorhodopsin: Primary proton transfer”, J. Phys. Chem. B, 124, 8524 (2020).
<LIB/SIB/PIB>
- M. Okoshi, C.-P. Chou, H. Nakai, “Theoretical analysis of carrier ion diffusion in superconcentrated electrolyte solutions for sodium-ion batteries”, J. Phys. Chem. B, 122, 2600 (2018).
- K. Doi, Y. Yamada, M. Okoshi, J. Ono, C-P. Chou, H. Nakai, A. Yamada, “Reversible sodium metal electrodes: Is fluorine an essential interphasial component?”, Angew. Chem. Int. Ed., 58, 8024 (2019).
- Q. Zheng, S. Miura, S. Ko, K. Miyazaki, E. Watanabe, M. Okoshi, C.-P. Chou, Y. Nishimura, H. Nakai, T. Kamiya, T. Honda, J. Akikusa, Y. Yamada, A. Yamada, “Sodium- and potassium-hydrate melts containing asymmetric imide anions for high-voltage aqueous batteries”, Angew. Chem. Int. Ed., 58, 14202 (2019).
<TDDFTB-MD>
- N. Komoto, T. Yoshikawa, J. Ono, Y. Nishimura, H. Nakai, “Development of large-scale excited-state calculations based on the divide-and-conquer time-dependent density functional tight-binding method”, J. Chem. Theory Comput., 15,1719 (2019).
- N. Komoto, T. Yoshikawa, Y. Nishimura, H. Nakai, “Large-scale molecular dynamics simulation for ground and excited states based on divide-and-conquer long-range corrected density functional tight-binding method”, J. Chem. Theory Comput., 16, 2369 (2020).
<SF-TDDFTB>
- M. Inamori, T. Yoshikawa, Y. Ikabata, Y. Nishimura, H. Nakai, “Spin-flip time-dependent density-functional tight-binding method for application to conical intersection”, J. Comput. Chem., 41, 1538 (2020).
<TDDFTB-FSSH>
- H. Uratani, H. Nakai, “Non-adiabatic molecular dynamics with divide-and-conquer type large-scale excited state calculations”, J. Chem. Phys., 152, 224109 (2020).
- H. Uratani, T. Morioka, T. Yoshikawa, H. Nakai, “Fast nonadiabatic molecular dynamics via spin-flip time-dependent density-functional tight-binding approach: Application to nonradiative relaxation of tetraphenylethylene with locked rings”, J. Chem. Theory Comput., 16, 7299 (2020).
- H. Uratani, T. Yoshikawa, H. Nakai, “Trajectory surface hopping approach to condensed-phase nonradiative relaxation dynamics using divide-and-conquer type spin-flip time-dependent density-functional tight-binding”, J. Chem. Theory Comput., 17, 1290 (2021).
<Perovskite Solar Cell>
- H. Uratani, C-P. Chou, H. Nakai, “Quantum mechanical molecular dynamics simulations of polaron formation in methylammonium lead iodide perovskite”, Phys. Chem. Chem. Phys., 22, 97 (2020).
- H. Uratani, H. Nakai, “Simulating the coupled structural–electronic dynamics of photo-excited lead iodide perovskites”, J. Phys. Chem. Lett., 11, 4448 (2020).
<Review>
- A. Sakti, Y. Nishimura, H. Nakai, “Recent Advances in Quantum-Mechanical Molecular Dynamics Simulations of Proton Transfer Mechanism in Various Water-Based Environments”, WIREs Comput. Mol. Sci., 10, e1419 (2019).
- C. Chou, A. Sakti, Y. Nishimura, H. Nakai, “Development of divide-and-conquer density-functional tight-binding method for theoretical research on Li-ion battery”, Chem. Rec., 18, 746 (2019).
<Review (in Japanese)>
- 西村 好史, 吉川 武司, 中井 浩巳, “DCDFTBMDプログラムの公開”, J. Comput. Chem. Jpn., 17, A21 (2018).