NOMO法の開発
1. NOMO法とは?
2. 理論
-2.1 NOMO/HF法
-2.2 NOMO/CIS法
3. 計算結果-NOMO/CIS法-
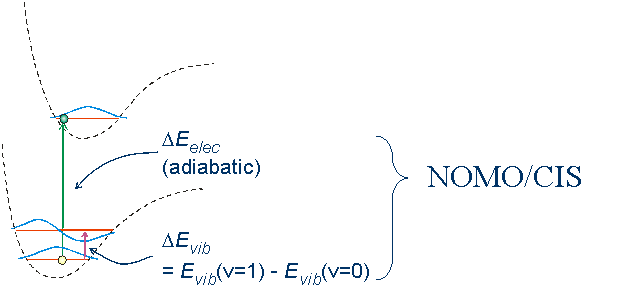
-3.1 電子および振動励起状態
-3.2 全エネルギーの原子核基底関数依存性
-3.3 差原子核密度図
NOMO法とは?
|
BornとOppenheimerによって提案された電子と原子核の運動を分離して扱うBorn-Oppenheimer(BO)近似は,分子軌道(MO)法や密度汎関数理論(DFT)などの電子状態理論の基礎となっている。BO近似のもとでは,ある特定の原子核配置に対する電子波動関数は,電子Hamiltonianに対する時間に依存しないSchrodinger方程式を解くことで求めることができる。近年では,電子状態計算の高精度化と高効率化が進み,その対象は小さな有機・無機分子に限らずナノ・バイオなどの大規模分子にも拡がりつつある。しかしBO近似では,ゼロ点振動・トンネル効果・共鳴・散乱・干渉など原子核の波動性に由来する現象の記述が困難である。この問題を解決するためには,原子核を量子論的に取り扱う必要がある。 |
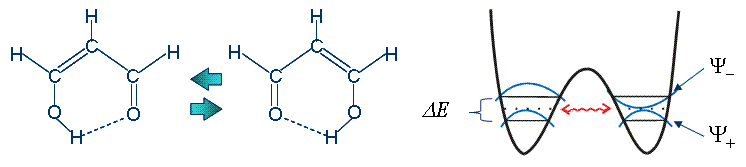
Figure 1. Proton tunneling in Malonaldehyde
|
そこで,我々のグループではnon-BO理論の開発に取り組んできた。我々の手法における重要な出発点は,原子核の1粒子軌道として核軌道(NO)を導入することである。これにより,従来のMO法をnon-BO問題へ拡張することができる。Nuclear orbital plus molecularorbital(NOMO)法と呼ばれる本手法は,BO近似を用いずに原子核と電子の波動関数を同時に決定することができる。原理的には,NOMO法は厳密解を与えることができる。近年,non-BO問題に対する同様のアプローチは他の研究グループによっても報告され,徐々にではあるが注目されつつある。NOMO法の実用的な発展として,まず一体近似のもとで原子核と電子の波動関数を決定するHartree-Fock(HF)法を開発してきた。またNOMO法の拡張として,CIS法,MP2法の開発を行ってきた。ここでは,NOMO/HF法の理論,NOMO/CIS法の理論および計算結果を示す。
理論
NOMO/HF法
NOMO/HF法ではHF波動関数を,電子波動関数と原子核波動関数の直積として表す。
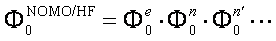
ここで1粒子波動関数(分子軌道(MO),原子核軌道(NO))を導入し,電子波動関数と原子核波動関数をそれらの反対称(対称)積として表す。
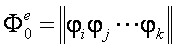
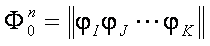
MO

NO
この波動関数を用いた全エネルギーをMO,NOについて変分することで,NOMO/HF方程式が導出される。
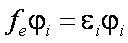
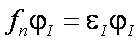
Fock演算子は,平均場的なNOとMO間の相互作用を含む。これは非制限HF(UHF)形式と類似の表式であり,MO/HF法からのNOMO/HF法への拡張は直接的であると言える。実際,NOMO/HF波動関数はKoopmansの定理やBrillouinの定理を満たす。したがって,BO近似のもとで発展してきた様々な相関手法をNOMO法に適用することができる。
NOMO/CIS法
NOMO/HF法は,基底状態における原子核と電子の波動関数を同時に決定することができる。ここではNOMO法を励起状態へと拡張することを試みる。NOMO/FCI手法は完全系を用いることで,基底状態及び励起状態に対して原理的には厳密な原子核と電子の波動関数を与える。従来のMO法において,励起状態に対する簡便な手法としては1粒子励起演算子を用いたCI(CIS)法がある。CIS法をNOMO法へと拡張することによって,原子核と電子の励起状態を与えることが期待される。
|
MO theory(BO approximation)

|
NOMO theory(non-BO)
|
Figure 2. Schematic pictures of MO and NOMO theories.
NOMO/CIS波動関数は以下のように与えられる。

CI係数は変分的,すなわち,CI行列の対角化によって決定される。
計算結果-NOMO/CIS法-
電子および振動励起状態
表1は,様々な原子核基底関数(NBF)を用いてNOMO/CIS法で計算されたH2,D2,T2 分子の振動(v=0→1,0→2)及び電子励起状態(S0→S1)のエネルギーを示す。電子基底関数(EBF)はcc-pVTZを用いた。比較として,実験値を示した。従来のMO/CIS 法では振動励起状態エネルギーを求めることはできない。
Table 1. Vibrational and electronic excitation energies (in cm-1) of H2, D2, and T2 calculated by the NOMO/CIS method, comparing with experiments.

全エネルギーの原子核基底関数依存性
図3には様々なNBFを用いたNOMO/HFとCIS計算によって得られたH2の基底状態と振動励起状態(v=0,1,2)のエネルギー差を示す。EBFはcc-pVTZを用いた。これらの状態に対する全エネルギーはNBFの増加と共に減少するが,基底状態エネルギーの依存性は小さいことがわかる。第1励起状態(v=1)の全エネルギーはp型関数を加えることで収束しているように思われる。第2励起状態(v=2)はd型関数を加えるまで収束していない。この傾向は第1,2励起状態の調和振動子の厳密な波動関数に対応する。
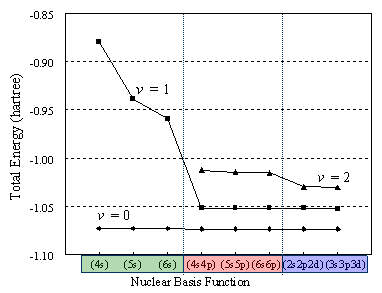
Figure 3. NBF dependence of the total energies of H2 in the ground and vibrational excited sates (v=0,1,and 2) calculated by the NOMO/HF and CIS methods.
差原子核密度図
図4にNOMO/CIS法によって得られたH2,HeH+,LiHの振動基底と励起状態間(v=1と2)の差原子核密度を示す。NBFとEBFとしては(3s3p3d)とcc-pVTZを用いた。pzとdzz2型関数の重要性が密度図に直接的に見られる。HからHe更にLiへと質量が増加するにしたがって,原子核密度は縮む。これは質量の増加と共に量子効果が減少することを意味している。HeH+における密度は他のものよりも広がっている。これはHが弱い化学結合によって広く振動していることを示している。
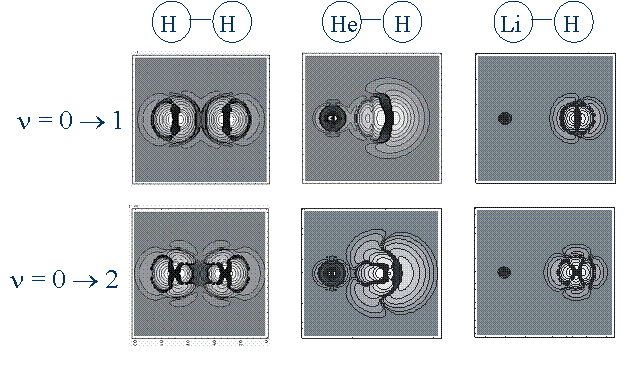
Figure 4. Nuclear density difference maps for the vibrational excitations (v=0→1 and 0→2) of H2, HeH+, and LiH.
次に,(4s4p4d)NBFを用いてNOMO/CIS法によりH3+の振動励起状態を見積った。基底状態構造におけるH3+の点群はD3hである。2つの最低状態はE’対称性を持って縮重している。第3励起状態はA1’対称性を持つ。これらの状態は反対称や全対称伸縮振動の励起状態に帰属される。図5(a),(b),(c)にはp型関数の方向を示した基底状態と3つの励起状態間の差原子核密度図を示す。これらの差密度は図5(d),(e),(f)に示すように振動モードと一致する。したがって,NOMO/CIS法は振動波動関数を適切に記述することがわかった。
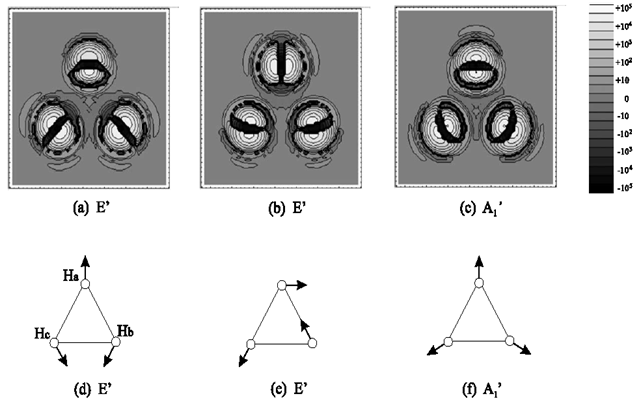
Figure 5. Nuclear density difference maps for the three vibrational excitations of H3+ (a)-(c) and schematic illustration for the three normalmodes of H3+ (d)-(f).
Topへ
研究紹介へ
中井件HOMEへ