メチル基内部回転運動のメカニズム
[1-4,8]
- π*-σ* Hyperconjugation mechanism on the rotational
barrier of the methyl group -
1. 序
2. 回転障壁の再現とメチル基回転のポテンシャルカーブ
3. メカニズムの解明 -π*-σ* Hyperconjugation mechanism-
4. 理論的予測
5. 発展
p
1. 序
フルオロトルエンなど、置換トルエン(Fig. 1)に付加したメチル基の、基底状態及び励起状態における内部回転障壁が、supersonic
jet技術により測定され、その回転障壁が励起により大きく変化する事実に注目が集まった[5]。Table
1に測定された置換トルエンの回転障壁を示す。オルト体は、その立体障害から基底状態において回転障壁が大きいが、励起状態では障壁が小さくなっている。また、表は置換基の電子供与性が大きい順にならべてあるが、供与性の強さとも相関がある。またメタ体はオルト体と逆の挙動を示している。その変化の原因は長らく謎であった。これに対して、分子軌道法を用いて理論的アプローチを試みる。
Figure 1. o-fluorotoluene
Table 1. Rotational barriers of the methyl
group in o- and m- substituted toluenes in cm-1.
| Substituent |
Species |
S0 |
◆ |
S1 |
◆ |
S1<-S0 |
◆ |
| ortho-system |
◆ |
exptl. [6] |
calc. [2] |
exptl. [6] |
calc. [2] |
exptl. [6] |
calc. [2] |
| NH2 |
o-toluidine |
703 |
665 |
40 |
72 |
-663 |
-593 |
| OH |
o-cis-cresol |
600 |
629 |
90 |
126 |
-510 |
-603 |
| OH |
o-trans-cresol |
355 |
366 |
-83 |
-123 |
-438 |
-489 |
| F |
o-fluorotoluene |
228 |
199 |
-22 |
-81 |
-250 |
-280 |
| CN |
o-tolunitrile |
-- |
197 |
-- |
714 |
-- |
517 |
| CH=CH2 |
o-tran-methylstyrene |
-- |
700 |
-- |
1224 |
-- |
524 |
| meta-system |
◆ |
◆ |
◆ |
◆ |
◆ |
◆ |
◆ |
| NH2 |
m-toluidine |
9 |
9 |
317 |
359 |
308 |
350 |
| OH |
m-cis-cresol |
11 |
-12 |
213 |
241 |
202 |
253 |
| OH |
m-trans-cresol |
26 |
37 |
211 |
268 |
185 |
231 |
| F |
m-fluorotoluene |
16 |
13 |
124 |
176 |
108 |
163 |
| CN |
m-tolunitrile |
14 |
-17 |
-39 |
-465 |
-53 |
-448 |
| CH=CH2 |
m-trans-methylstyrene |
52 |
15 |
-79 |
-395 |
-131 |
-410 |
2. 回転障壁の再現とメチル基回転のポテンシャルカーブ
Table 1に本研究による回転障壁の計算値を示す。計算レベルはHartree
Fock及びCIS法を用い、基底関数は6-31G**である。電子相関効果が入っていないにも関わらず、計算値は実験値をよく再現している。(これは、メチル基の回転に対しては電子相関効果があまり効かないからである。)
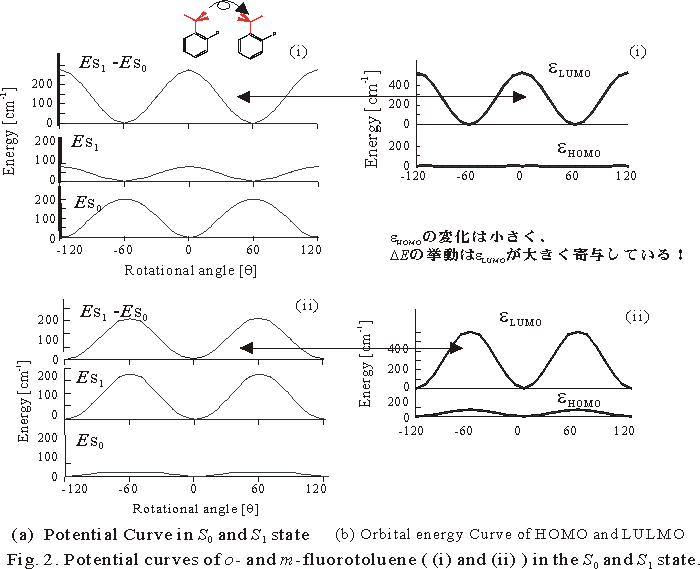
ここで、励起に伴う回転障壁変化に注目してみよう。これは、ほぼ励起エネルギーのメチル基回転に対する変化に対応している。回転障壁変化は、電子供与性もしくは吸引性が大きいほど変化の絶対値が大きく、オルト体とメタ体で挙動が逆であることが分かる。
これら置換トルエン系の第一励起状態は、LUMO<-HOMO励起が主配置となる。励起エネルギー(fig.2/上段のカーブ)は、第一近似的にはHOMOとLUMOの軌道エネルギー差で表すことができる。そこで、HOMOとLUMOの軌道エネルギーの挙動を見てみると、HOMOの軌道エネルギーは、メチル基の回転によってほとんど変化せず、LUMOの軌道エネルギーの変化が励起エネルギーの挙動とよく対応していることが分かる(seeFig.
2 (b))。LUMOの軌道エネルギーの挙動が回転障壁の変化の原因でありそうだ。そこで、LUMOの変化の原因が何であるか調べてみよう。
3. メカニズムの解明 -π*-σ* Hyperconjugation mechanism-
それでは、LUMOのMO図を見てみよう。(fig.3) 数字はMO係数である。オルト体においては60°体のメチル基面外水素の係数が大きくなっていることが分かる。空間的に近いオルト炭素原子(メチル基の結合している炭素の隣の炭素)と位相が一致しているために、水素に非局在化しているのだ。反対側の配座では、位相が異なる(この場合は、電子密度が無い)ために、安定化はおこらない。この違いにより、メチル基の回転によって、回転障壁が大きく変化するのである。
また、LUMOの形は、置換基の結合する位置によっても変わる。フッ素は電子供与性であるために、結合する炭素の電子密度が少ないほうが安定となる。そのため、メタ体では形が変わり(回転し)、オルト体と逆の0°体で超共役が起こる。このことが、回転障壁変化のオルト-メタの逆転効果を引き起こしているのである。
この非局在化は、有機化学では有名な超共役によく似ている。普通の超共役が、メチル基のσ(擬π)軌道と、メチル基が結合した炭素との間の相互作用である(π-σ)のに対し、この相互作用は、メチル基のσ*(擬π*)軌道とオルト炭素との相互作用であるため、われわれはこの相互作用をπ*-σ*-Hyperconjugationと名づけた。この相互作用は、Virtual(仮想)軌道での相互作用であるため、励起状態でしか効果を表さず、また、オルト炭素との相互作用であるため、回転障壁に対して大きく寄与するという特徴がある。
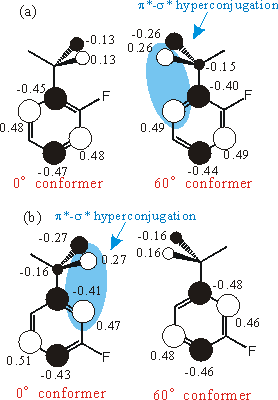
Fig. 3 LUMO pictures of o-and m-fluorotoluene ((a) and (b)).
4. 理論的予測
それでは、次に、LUMO+1軌道を見てみよう。(fig.4)LUMOとは形が異なるので、LUMOとちょうど逆の配座で超共役が起こっているのが分かる。このことから、LUMO+1への励起が主配置となる(実験的に測定されていない)第二励起状態では、回転障壁変化が逆転することが理論的に予測される。
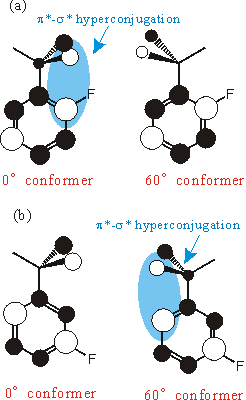
Fig. 4 LUMO+1 picture of o- and m-fluorotoluene ((a) and (b)).
ところで、これらLUMO、LUMO+1軌道は、置換基の結合していないベンゼンでは縮重している。このとき、第一励起状態では、ちょうどLUMOへのLUMO+1が1:1でミックスしている。置換基がつくことによってその縮重は解けるのであるが、このとき、電子吸引性か、供与性かで、どちらがLUMOになるかが決定する(供与性基はFluorotolueneにおけるLUMO、吸引性基はLUMO+1を安定化させるため)。またその分裂幅は、供与、吸引性が強いほど大きく分裂する。ここで、これら二つのLUMOがπ*-σ*超共役において、逆の寄与を起こすことに注意すると、分裂幅が小さいと、互いの効果がキャンセレーションを起こし、回転障壁変化が小さくなり、逆に分裂幅が大きいと、回転障壁変化が大きくなるということに気づく。つまり、置換基の電子供与性、吸引性が大きいほど回転障壁変化は大きく、逆に小さいほど回転障壁変化も小さくなる。
まとめると、
(1) オルト体 v.s. メタ体
(2) 第一励起状態 v.s. 第二励起状態
(3) 電子供与性 v.s. 電子吸引性
という三つのプロパティに対して、回転障壁の逆転効果が予測されることになる。
Fig. 5に、第一及び第二励起による回転障壁変化を、Hammettのσに対してプロットした。Hammett
σは、有機化学でよく用いられる電子供与、吸引性の指標となる値である。先に述べた三つの逆転効果が実際に現れていることが分かる。
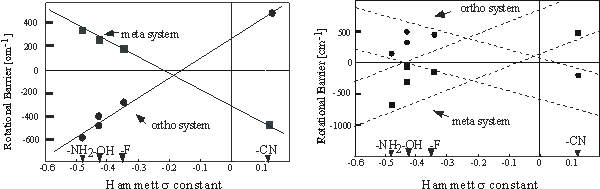
Fig. 5 Correlation between Hammett σ constant and the barrier change
by S1-S0 (left) and S2-S0 (right) excitation.
また、アニオン化状態について考えると、アニオン化は近似的にはLUMOにひとつ電子が入ると考えることができるが、HOMOの影響が小さいため、これは第一励起状態に対応すると考えることができる。アニオン化状態における回転障壁は、実験的に測定する方法がそもそも確立されていないが、もし測定できれば、それは第一励起状態と似た回転障壁変化を起こすと予測される。
このようなメカニズムの解明により、まだ実験的に測定されていない物質の回転障壁に対しても、理論的予測を行うことができる。これが理論研究の大きなメリットである。
5. 発展
今回は、置換トルエンについて述べたが、π*-σ*-Hyperconjugationメカニズムは、多くのメチル置換芳香族に対して適用できる。既に多環芳香族化合物[3]や、ヘテロ環化合物[4]について確認している。特に、多環化合物であるメチルナフタレンでは、HOMOにπ*-σ*-超共役が現れ、基底状態で大きな回転障壁を与えるなどの興味深い現象に対して、コンシステントな説明に成功している。
また、オルトトルニトリルなど、実験的にスペクトルが得られても、振動のピークと混ざって内部回転のピークが特定が困難な化合物に対して、理論的に得られたポテンシャルカーブからスペクトルを再現し、実験スペクトルの帰属にも成功している。[7]
References
[1] H. Nakai and M. Kawai, Chem. Phys. Lett. 307, 272
(1999)
[2] H. Nakai and M. Kawai, J. Chem. Phys. 113,
2168 (2000)
[3] H. Nakai and Y. Kawamura, Chem. Phys. Lett.
318, 298 (1999)
[4] Y. Kawamura, T. Nagasawa, H. Nakai, J. Chem.
Phys., 114, 8358 (2001)
[5] K. Okuyama, N. Mikami, and M. Ito, J. Phys.
Chem. 89, 5617 (1985)
[6] Z. Zhao, C. S. Parramenter, D. B. Moss, A.
J. Bradley, E. W. Knight, K. G. Owens, J. Chem. Phys. 96, 6362 (1992)
[7] 千葉、河村、中井、2000年分子構造総合討論会(東京)
[8] Y. Kawamura, H. Nakai, Chem. Phys. Lett. 368,
673 (2003).
Topへ
研究紹介へ
中井件HOMEへ
◆2001.8 Y.Kawamura