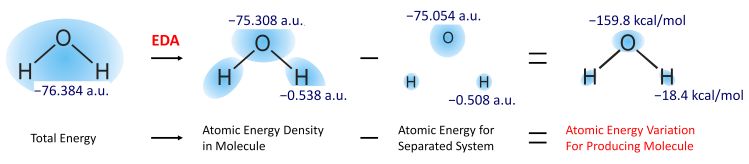
Figure 1. The energy densities and the energy density changes of oxygen and hydrogen atoms on water molecule.
近年、電子状態理論計算は高精度で合理的な結果を導出するため注目を集めるようになった。たとえば固体表面への吸着現象は触媒反応や電極反応のメカニズムとして、有機化学反応は生体内反応の構成要素としてそれぞれ興味深く、電子状態計算により研究され、化学的に理解することが試みられてきた。しかし、理論計算から得られる情報は計算コストなどの制約によって限られており、優れた解析手法の開発は、電子状態理論計算の結果からより多くの化学的理解を得るために重要である。
Mullikenの電子密度解析(MPA)は分子内の電子を構成原子ごとに分割する解析手法であり、原子価状態などを解析する方法として非常に広く用いられている。当研究室では、密度汎関数法(DFT)により得られた全エネルギーを構成原子ごとに分割する解析手法、エネルギー密度解析(EDA)[42]を開発した。ここで、EDAを水分子に対して適用した例をFigure 1に示す。この例では計算によって得られた全エネルギーを酸素原子と水素原子に対して分割している。これらの値と原子単体時のエネルギーとの差を計算することにより、原子化エネルギーに対するそれぞれの原子の寄与を計算することができる。
Figure 1. The energy densities and the energy density changes of oxygen and hydrogen atoms on water molecule.
EDAを様々な系に適用することにより、表面吸着現象の解明[57,75]や新しい有機化合物の設計[65]などの、多くの優れた成果を挙げてきた。ここでは、Si表面への吸着現象に対する解析を示す。
固体表面への分子吸着は触媒反応の初期過程として重要であり、電子状態理論の発展と計算機能力の向上により、これらの系は電子状態計算でもよく取り扱われるようになった。電子状態計算で固体表面を取り扱う場合、半無限に広がる表面に対してなんらかのモデル化が必要となる。表面反応の局所性を仮定して、有限個の原子を切り出すクラスターモデルはよく用いられているが、実際に反応の局所性を評価することは不可能であり、どんな大きさ・形のクラスターを用いればよいかの指針は無かった。
共有結合性結晶であるSiクラスターに対しては、クラスターの端のSi原子をH原子などでキャップするcapped bond modelがよく用いられる。Si45H36クラスターに対するH2、C2H2、C2H4吸着によるエネルギー密度変化をFigure 2に示す[57]。表面第1層の原子について、吸着サイトであるAではその他の原子と比べて大きく安定化しているが、隣のサイトBでは吸着によるエネルギー密度変化が2 kcal/mol程度になり、サイトCでは1 kcal/mol以下になっている。また深さ方向に対しては、3層目ではサイトAでも2 kcal/mol以下になっており、相互作用が収束していることがわかる。EDAを用いることにより、相互作用の局所性を評価可能にし、どのような大きさ・形のクラスターを用いればよいかの指標を得ることに成功した。
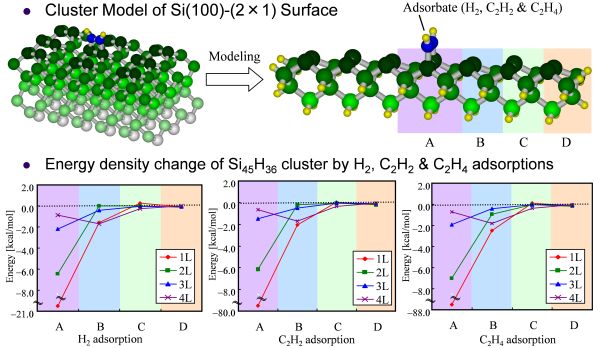
Figure 2. Energy density changes of Si atoms by H2, C2H2, and C2H4 adsorption on Si45H36 cluster (B3LYP/cc-pVDZ).
EDAの有用性をさらに増加させるための拡張として、Interaction-EDA[58]、Bond-EDA[62]、ETS[67]、NAO-EDA[82]、Grid-EDA[89]、PBC-EDA[94]などが開発されてきた。ここでは、そのいくつかについて解説する。
分子間相互作用の詳細な理解のために、Kitaura–Morokuma法や制限変分空間自己無撞着(RVS-SCF)法など、量子化学計算から得られる相互作用エネルギーを静電(ES)・交換(EX)・分極(PL)・電荷移動(CT)などの物理的に意味のあるいくつかの成分に分割するエネルギー分割法が古くから用いられてきた。しかし、電子状態計算で取り扱う系は近年1000原子を超えるような大規模系を対象としており、そのような大規模分子間の相互作用においては、系内のどの原子、どの置換基が相互作用に寄与しているかを特定することは困難であった。その解析を目的として、エネルギー分割法の一つであるRVS-SCF法とEDAとをFigure 3に示すように組み合わせることで、相互作用エネルギー成分を局所領域に分割する解析手法、Interaction-EDA[58]を開発した。
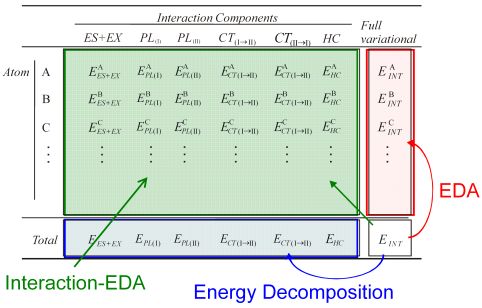
Figure 3. Partitioning scheme of Interaction-EDA.
分子内の化学結合の理解に対する要求は古くからあり、化学的・物理学的に非常に重要である。化学結合は直接的には測定されない概念であり、種々の実験的・理論的な方法を用いてその解析が行われてきた。化学結合や分子間相互作用をより詳細に解析する為、本研究室ではEDAの拡張として、電子状態計算により求められた分子のエネルギーを原子と結合領域に分割する解析手法、Bond-EDA[62]を開発した。本手法を用いることにより、Diels–Alder反応の解析[98]と、新しい超原子価化合物の設計[113]が行われた。ここでは、Diels–Alder反応(Figure 4)のメカニズム解析に対する適用例を示す。
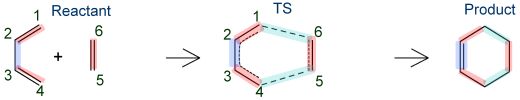
Figure 4. Scheme of Diels–Alder reaction.
ブタジエンとエチレンの環化付加反応は、量子化学的にもっとも基本的なDiels–Alder反応のうちの一つである。この反応のBond-EDAによる解析結果をTable 1に示す。この反応ではC1–C2、C3–C4、C5–C6の二重結合は単結合となるが、反応がすすむにつれて当該領域のエネルギーが不安定化し、生成物のエネルギーは反応物と比べて15–20 kcal/mol不安定化する描像が得られた。一方、C2–C3の単結合は二重結合となるが、当該領域でエネルギーが約4 kcal/mol安定化する描像が得られた。C1–C6、C4–C5の結合が生成する領域では、約120 kcal/molの安定化が得られた。
このようにして、化学反応における原子および原子間(結合領域)の相互作用をエネルギーとして見積もることに成功している。
Table 1. Bond lengths (R) and change of bond energy densities (ΔEbond) of Diels–Alder addition.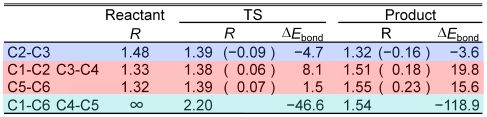
周期境界条件(PBC)計算は、単位構造を周期的に繰り返す無限系を表現することができる計算方法であり、固体表面・結晶モデルに対して理想的な計算方法である。しかし、固体表面上で起こる現象は局所的であるので、これを評価できる解析手法の開発が望まれた。そこでGauss基底を用いたPBC計算にEDAを組み合わせたPBC-EDA[94]を開発した。Figure 5に、PBC-EDAをMgO(110)、(100)表面に適用して得られたバルク中央層と表面の原子化エネルギーを示す。バルク中央層の原子化エネルギーは、それぞれ異なる表面であるにもかかわらず、層の数の増加に伴い凝集エネルギーの実験値(240 kcal/mol)に収束し、バルクを再現した。一方、表面の原子化エネルギーは(110)表面が213 kcal/mol、(100)表面が246 kcal/molと、それぞれバルクとは異なる値に収束し、(110)表面が(100)表面より不安定となった。これは、(110)表面では正(負)イオン同士が隣接するため、イオン結合のバランスが崩れて不安定化したものと考えられる。2次元PBC-EDAを用いることで、表面とバルクの安定性を同時に評価できるので、深さ方向に注意すれば、PBC計算における理想的な固体表面モデルの設計を行うことができることが示唆された。
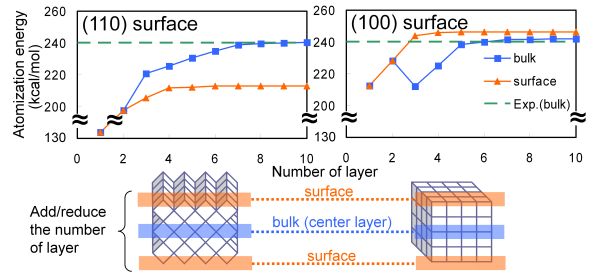
Figure 5. Atomization energies of MgO(110) and MgO(100) surfaces with respect to the number of layers (BLYP/6-21G).